HIJACKED POWERHOUSES: HOW CANCER USES MITOCHONDRIA TO WEAKEN IMMUNE DEFENSES
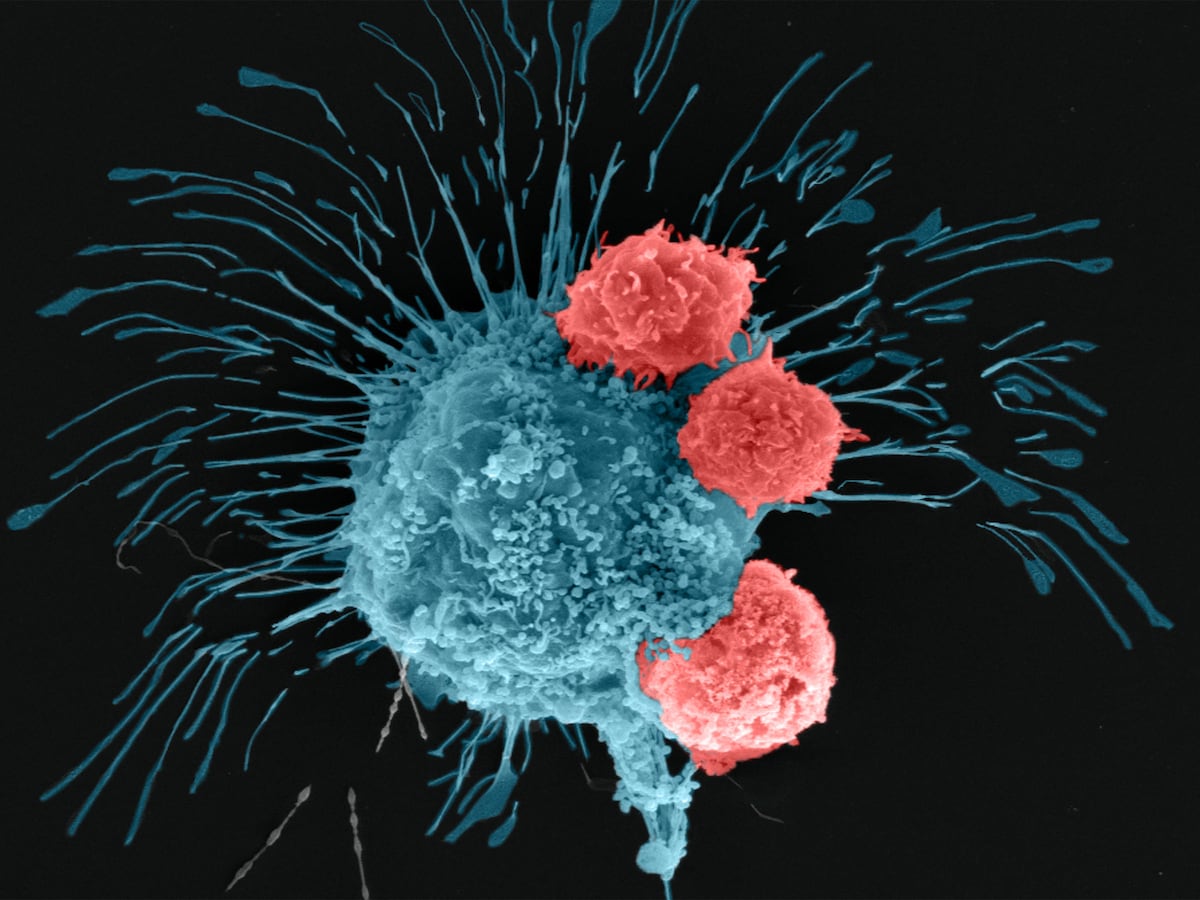
Cancer is a formidable disease that arises from uncontrolled cell growth, often defying the body's natural defense mechanisms. It employs sophisticated immune evasion strategies, known as immunoediting, to prevent detection and destruction. Initially, the immune system recognizes and eliminates tumor cells, but genetic instability enables cancer variants with reduced immunogenicity to persist and grow. Tumors also suppress immune responses by inducing CD4+ CD25+ FoxP3+ regulatory T cells (Tregs), which promote an immunosuppressive environment, myeloid-derived suppressor cells, M2 macrophages, and modulated dendritic cells that create a pro-inflammatory environment that promotes tumor progression. Additionally, downregulation of antigen processing machinery, secretion of immunosuppressive cytokines (e.g., TGF-β, IL-10, TNF-α), and expression of inhibitory immune-checkpoint molecules (e.g., CTLA-4, PD-1, PD-L1) further enable immune escape. These mechanisms present significant challenges for conventional therapies like chemotherapy and radiotherapy.
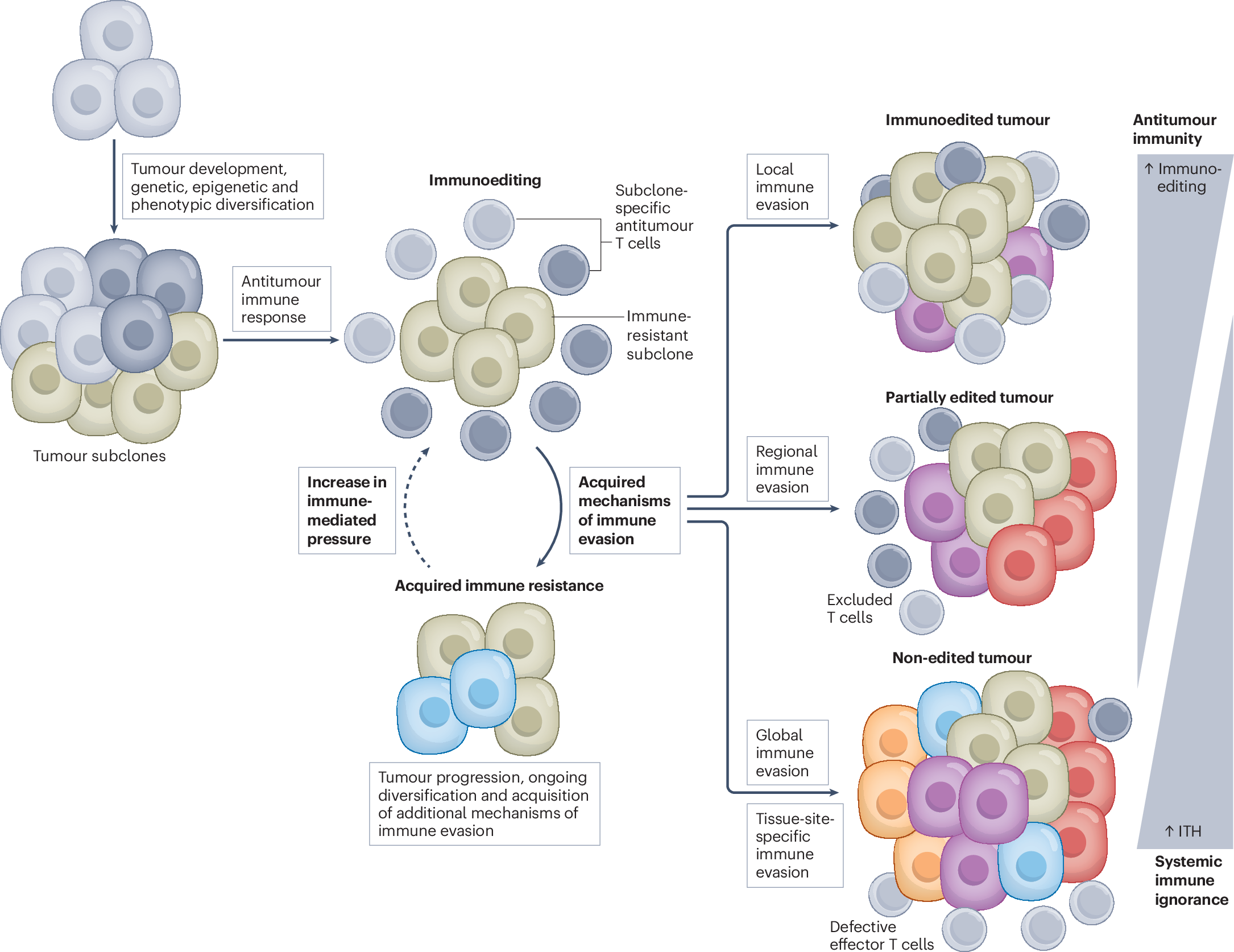 |
| Process of immunoediting |
Immunotherapies, especially immune checkpoint inhibitors (ICI) treatment, which includes anti-PD-1, anti-PD-L1, and anti-CTLA-4 monoclonal antibodies, have been gaining momentum but also faced challenges such as failure to obtain desirable responses progressively despite a good initial response. Thus, identification and understanding of various immune evasion mechanisms are necessary to improve the efficiency of ICI treatment. Recently, scientists have identified a new mechanism by which cancer evades our immune system: mitochondrial transfer to tumor-infiltrating lymphocytes (TILs), especially T-cells. This transfer results in metabolic reprogramming, which affects the function of T-cells.
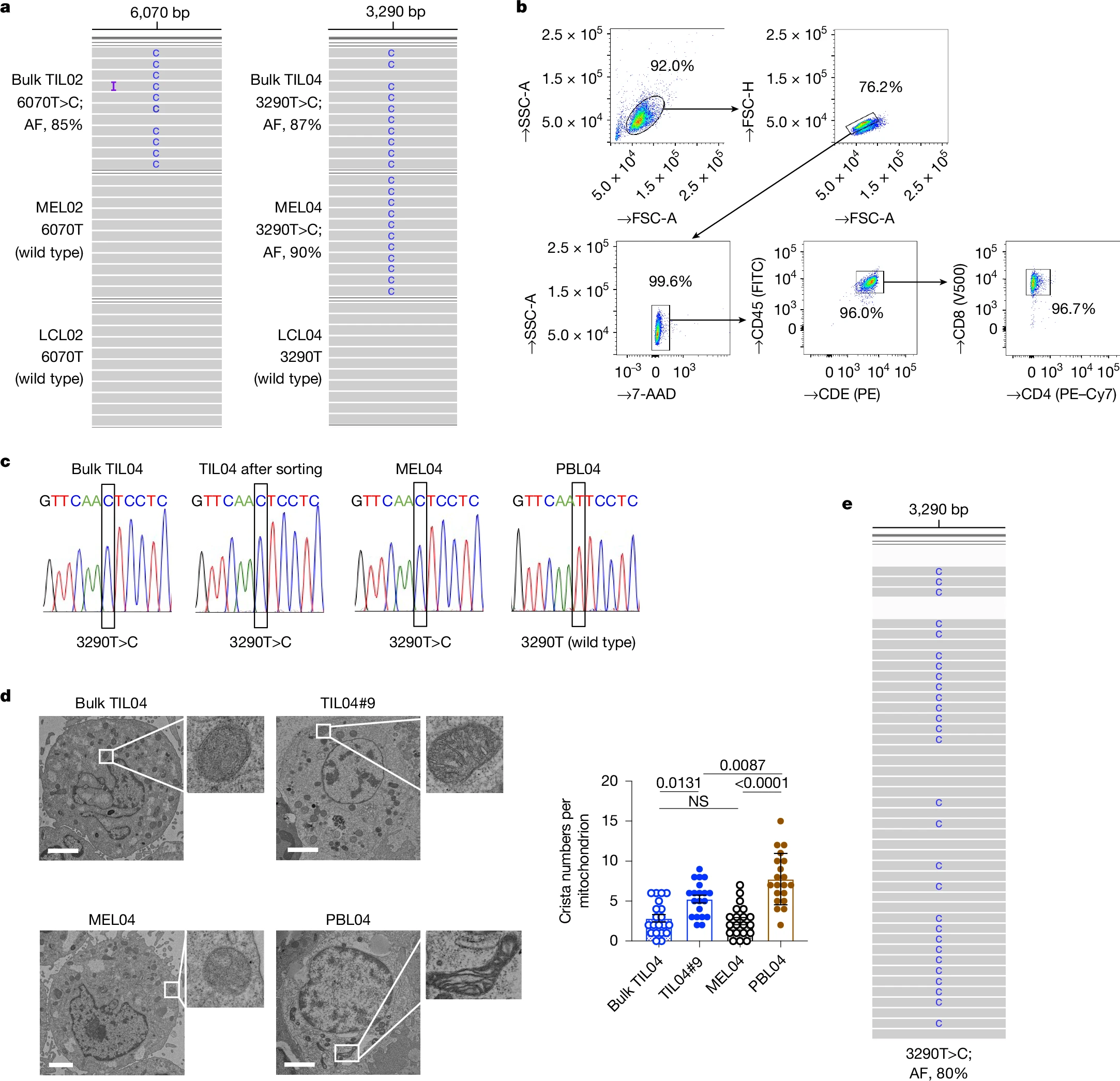
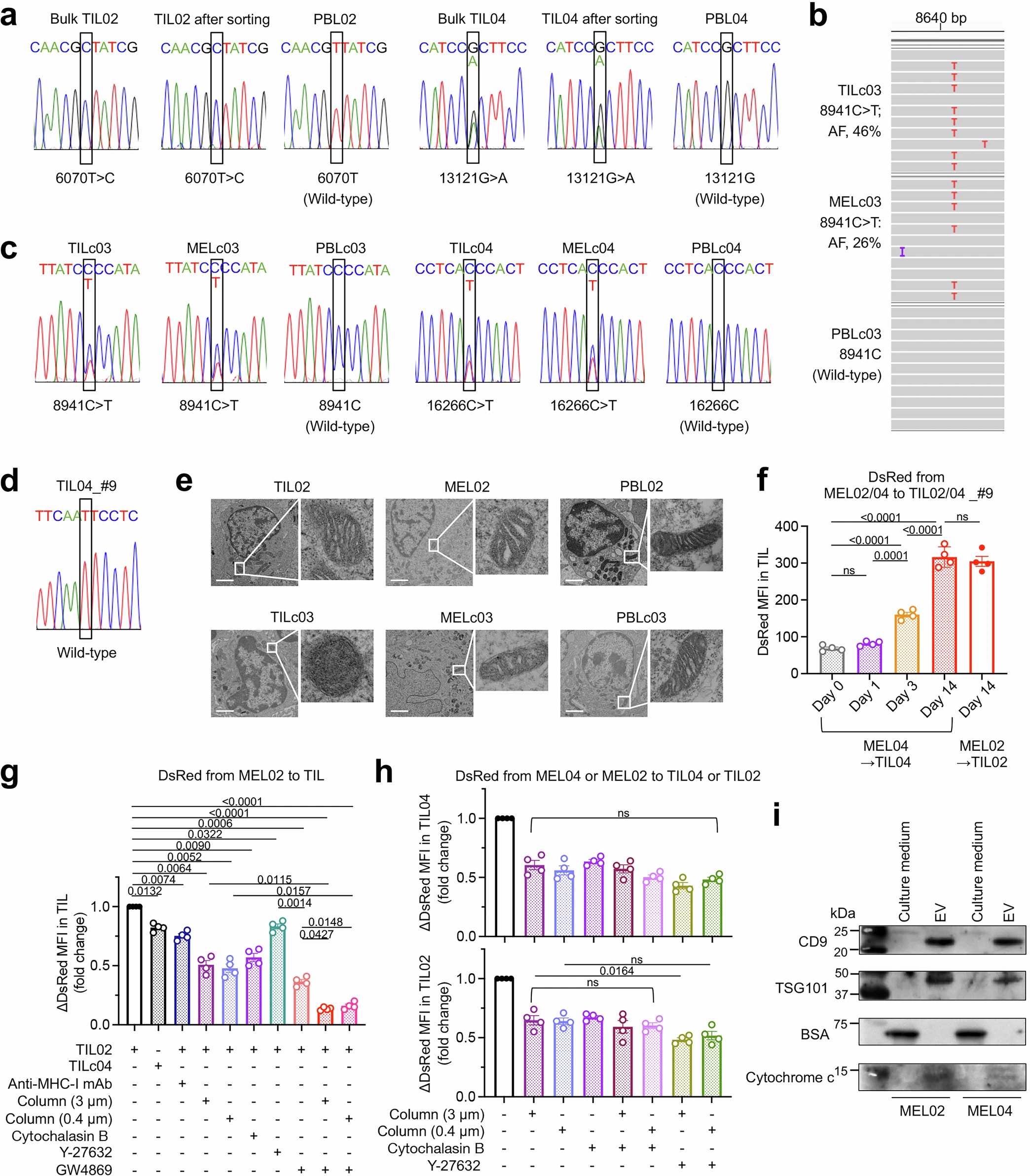
The sequencing of mitochondrial DNA (mtDNA) from TILs obtained from 12 patients with various cancers showed that 5 out of 12 patients had mtDNA mutations in TILs, which were predominantly CD45+ CD3+ T cells. A study on established cancer cell lines obtained from 7 patients obtained from who had 4 mtDNA mutations showed that 3 out of 4 mutations were shared between TILs and cancer. Electron microscopy revealed abnormal mitochondrial morphology with reduced cristae in both TILs and cancer cells having mtDNA mutations compared to TILs and cancer cells containing wild-type mtDNA.
.png)
.png)
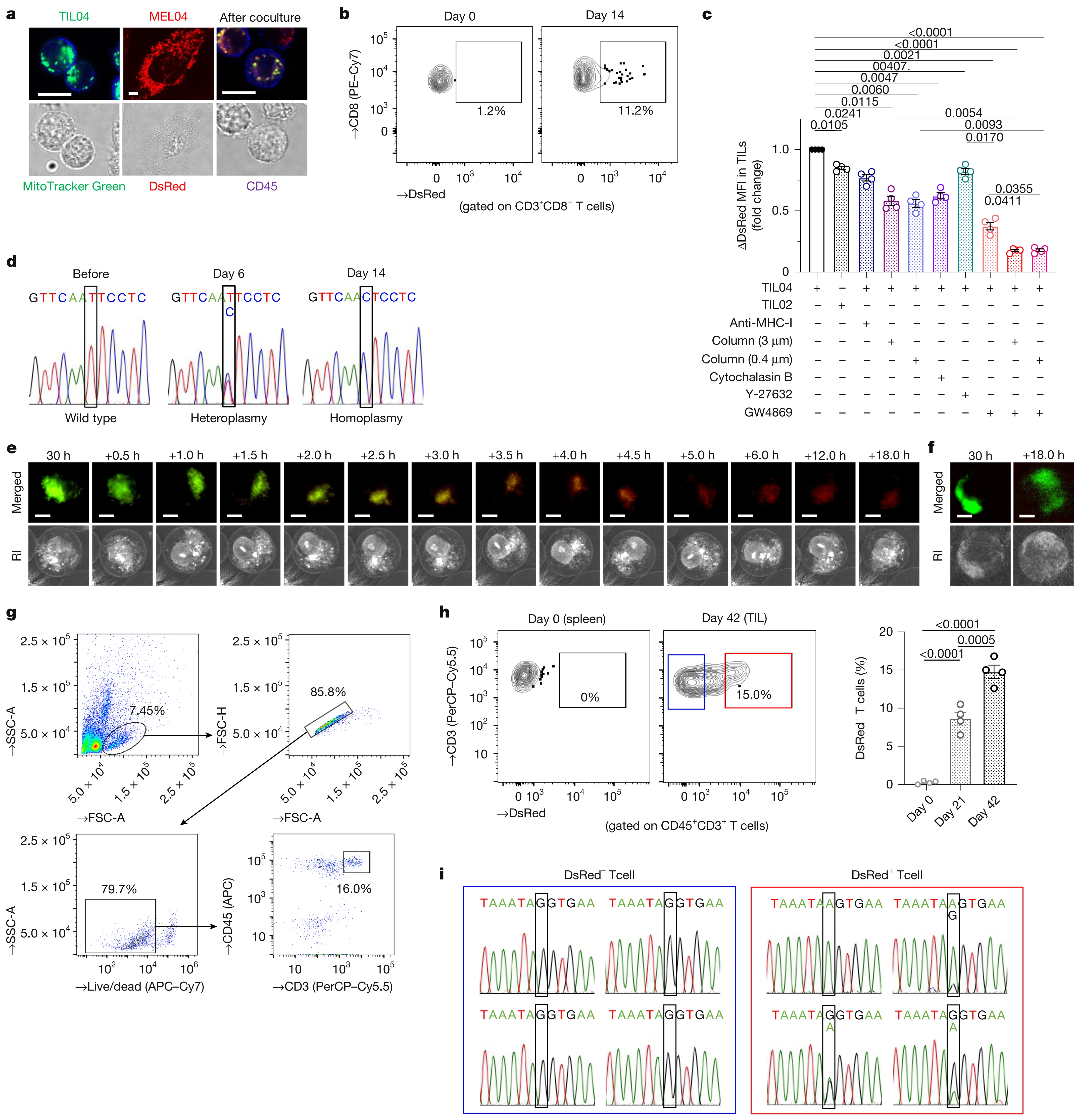
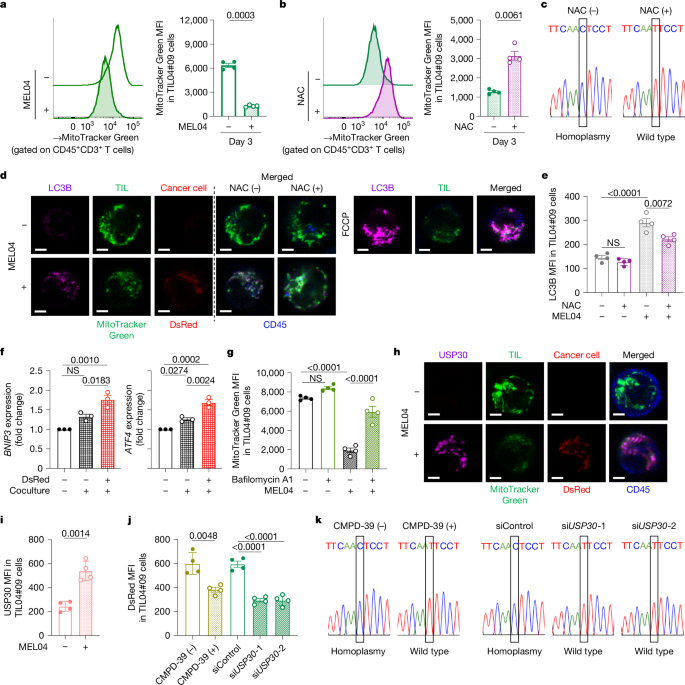
To confirm whether the observed mtDNA mutations shared between cancer and TILs may arise due to mitochondrial transfer, a mitochondria-specific fluorescent protein MitoDsRed was introduced into cancer cells containing wild-type mtDNA, named MEL02 cells, and containing mutated mtDNA, named MEL04. When these were co-cultured with TILs, mitochondrial transfer was observed to T-cells from cancer cells after 24 hours. The status of mtDNA did not affect the transfer efficiency. The formation of tunneling nanotubes (TNTs) and extracellular vesicles (EVs) has been identified previously as a mechanism of mitochondrial transfer in different cells. To test whether the same mechanisms are involved, cells were cultured in the presence of cytochalasin B, a TNT inhibitor, cell-culture column inserts (3 μm and 0.4 μm) to direct cell-to-cell contact, and GW4869, which inhibits the release of small EVs (~200 nm). Both resulted in a decrease in transfer to TILs. This showed that both direct cell-to-cell contact through tunneling nanotubes and transfer via extracellular vesicles are important transfer mechanisms. The EVs were isolated and analyzed, which revealed the presence of cytochrome C, CD9, TSG101, and small EV proteins, confirming that EVs contain mitochondria.
.png)
.png)
.png)
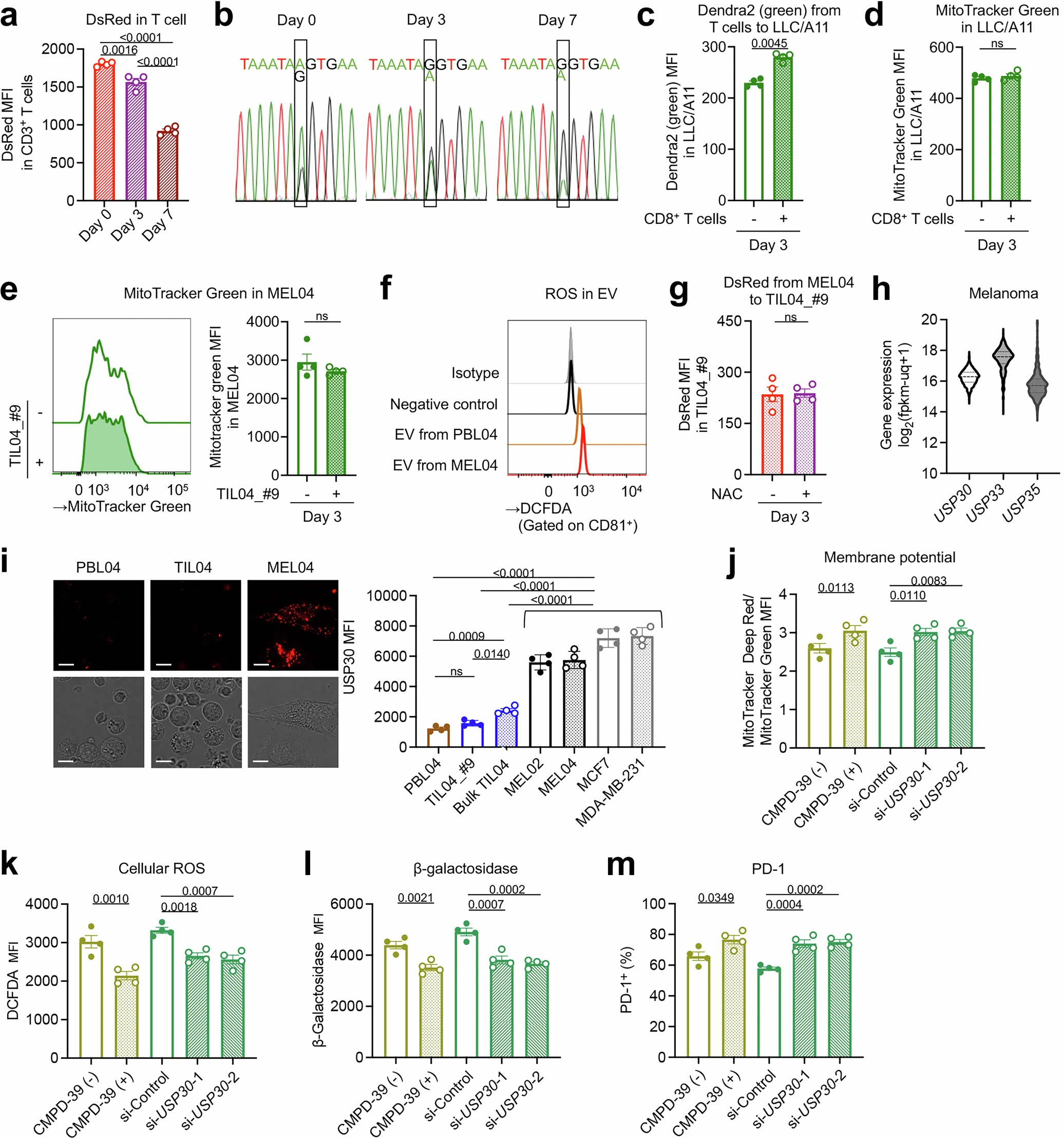
To study mitochondrial transfer from T-cells to cancer cells accurately, T-cells from PhaMexcised OT-1 mice, which express a mitochondrial-specific fluorescent protein (mito-Dendra2) with ovalbumin (OVA)-specific T-cell receptor, and OVA-overexpressing LLC/A11 cancer cells were co-cultured. The cancer cells were analyzed after 3 days using flow cytometry, and it was found that mito-Dendra2 expression had increased in LLC/A11 cells, indicating mitochondrial transfer from T-cells to cancer cells. It was also found that in-situ mitochondria in cancer cells were reduced while in-situ mitochondria in T-cells had decreased during co-culture, which was evaluated by MitoTrackerGreen.
.png)
Since it was known that reactive oxygen species (ROS) produced by cancer cause mitophagy (destruction of mitochondria) in neighboring cells, it was suspected that cancer induces a reduction in in situ mitochondria in TILs. To evaluate this, ROS was scavenged by N-acetyl-L-cysteine (NAC) treatment in co-culture, and it was found that the reduction of MitoTrackerGreen-labeled in-situ mitochondria in TILs and homoplasmy does not occur in NAC treatment. However, the treatment did not affect mitochondrial transfer from MitoDsRed-overexpressing cancer cells to T cells. LC3B staining confirmed that mitophagy was enhanced in TILs exposed to MitoDsRed-overexpressing cancer cells, but NAC treatment reversed this effect. Gene expression analysis further revealed upregulation of BNIP3 and ATF4, which are mitochondrial stress response genes. Bafilomycin A1, a mitophagy inhibitor, also showed prevention of the reduction of in situ mitochondria in T cells. These collectively suggested that mitochondria from cancer cells are more resistant to in situ mitochondria in T cells against mitophagy induced by ROS.


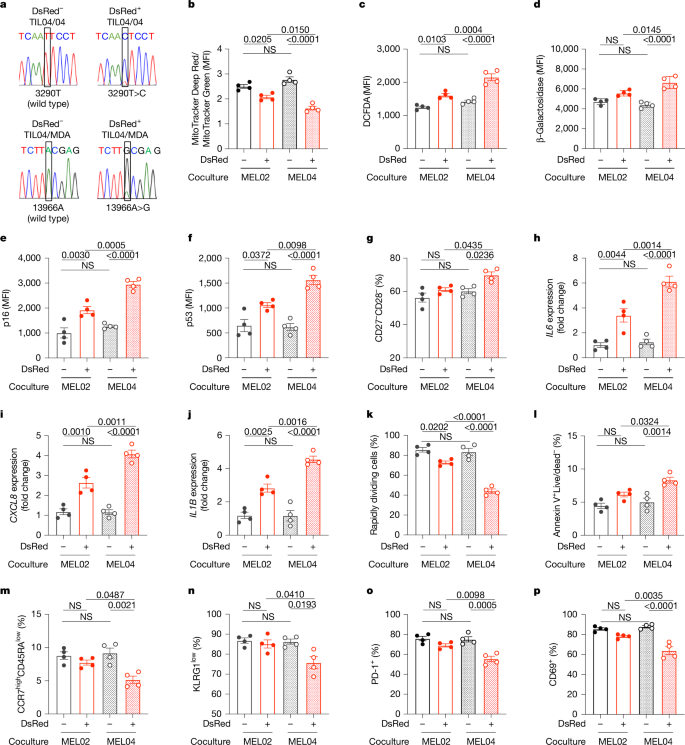
Then, the mitochondrial function was assessed in mutated T cells. It was observed that expression of mitochondrial molecules like ND4, ND5, ND6, CYTB, COX1, and ATP6 was repressed in mutated mitochondria, along with reduced basal respiration and increased dependence on glycolysis in mtDNA-mutated cancer cells. In case of CD8+ T cells co-cultured with cancer cells, it was observed that mtDNA-mutated TILs had a significant reduction in membrane potentials, substantial increase in ROS production, increased β-galactosidase activity, and levels of senescent molecules p16 and p53. Further analysis also revealed that mtDNA mutations impair memory formation and partially induce apoptosis in the central memory fraction. The activation of TILs was also found to be suppressed by the presence of mutated mitochondria.
 |
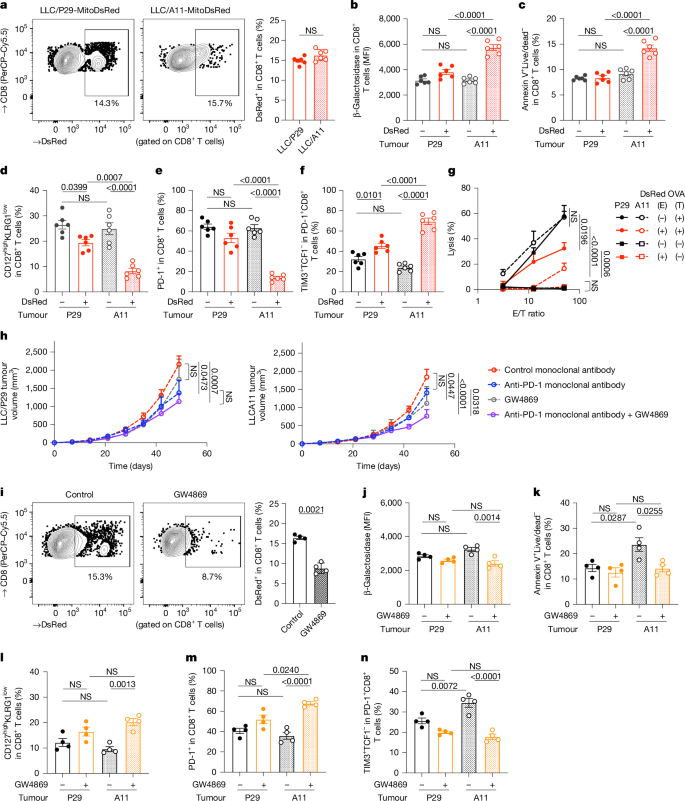
To explore how mitochondrial transfer affects T cells in vivo, researchers used two versions of Lewis lung carcinoma cells—LLC/P29, which has normal mtDNA, and LLC/A11, which carries a mutation in the Nd6 gene. LLC/A11 cells showed signs of mitochondrial dysfunction with reduced expression of ND6, impaired oxidative phosphorylation (OXPHOS), and a heavier reliance on glycolysis. The activity of mitochondrial complex I was also lower in LLC/A11 compared to the wild-type LLC/P29. Both cancer cell lines were engineered to express MitoDsRed. After 42 days of tumor growth in mice, it was found that both tumor types had a comparable level of CD8+ T cell infiltration, and the number of DsRed+ T cells (indicating mitochondrial transfer) was also similar between the two groups. However, in tumors derived from the mutated LLC/A11 line, DsRed+ T cells contained the same mtDNA mutation, confirming successful mitochondrial transfer. These T cells showed higher β-galactosidase activity, increased apoptosis, and a drop in MPECs (long-lived memory precursor effector cells). Furthermore, PD-1 expression, which normally signals T cell activation, was lower in these mutated T cells, suggesting that their activation was impaired due to mitochondrial dysfunction. These DsRed+CD8+ T cells from LLC/A11 tumors displayed a more exhausted phenotype, with higher TIM3 and lower TCF1 expression—hallmarks of terminally exhausted T cells. To assess functionality, the cytotoxicity of these T cells in a killing assay against OVA-expressing cancer cells was tested. CD8+ T cells that had received wild-type mitochondria from LLC/P29 showed a slight drop in killing power. But those with mutated mitochondria from LLC/A11 showed a marked loss of cytotoxic function. In mice, depleting CD8+ T cells sped up tumor growth in the LLC/P29 group—but had almost no effect in LLC/A11 tumors, reinforcing the idea that CD8+ T cells in LLC/A11 tumors were already dysfunctional. Interestingly, although LLC/P29 tumors grew faster in vitro, they were more sensitive to immune attack in vivo, showing a clear difference based on immune response. When the researchers applied PD-1 immune checkpoint therapy, only tumors with wild-type mitochondria (LLC/P29) responded. Tumors with mutated mitochondria (LLC/A11) were resistant to PD-1 blockade, and the therapy did not impact mitochondrial transfer at all. In essence, the transfer of mutated mitochondria from cancer cells cripples CD8+ T cells, pushing them into dysfunction and exhaustion, and undercutting the effectiveness of immunotherapy.

In conclusion, cancer’s ability to evade the immune system is a major barrier to effective treatment, and recent discoveries have revealed a novel mechanism behind this evasion involving the hijacking of mitochondria. These organelles, essential for cellular energy and immune function, are transferred from cancer cells to tumor-infiltrating lymphocytes (TILs), particularly T cells, leading to metabolic reprogramming and mitochondrial dysfunction in immune cells. This mitochondrial transfer impairs T cell activation, promotes senescence and exhaustion, and ultimately diminishes their ability to target and destroy tumor cells. The significance of this finding lies in its potential to explain why some tumors resist immunotherapy and highlights new targets, such as blocking mitochondrial transfer or restoring mitochondrial health in T cells, for improving cancer treatment outcomes.
REFERENCES:
Vinay, D. S., Ryan, E. P., Pawelec, G., Talib, W. H., Stagg, J., Elkord, E., Lichtor, T., Decker, W. K., Whelan, R. L., Kumara, H. M. C. S., Signori, E., Honoki, K., Georgakilas, A. G., Amin, A., Helferich, W. G., Boosani, C. S., Guha, G., Ciriolo, M. R., Chen, S., Mohammed, S. I., … Kwon, B. S. (2015). Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Seminars in cancer biology, 35 Suppl, S185–S198. https://doi.org/10.1016/j.semcancer.2015.03.004
Ikeda, H., Kawase, K., Nishi, T. et al. Immune evasion through mitochondrial transfer in the tumour microenvironment. Nature 638, 225–236 (2025). https://doi.org/10.1038/s41586-024-08439-0
IMAGE SOURCES:


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments
Post a Comment